LUMPAC USER's Guide
Module 1: Geometry Optimization
Geometry optimization is the first step to study the luminescence of a system containing a lanthanide ion. The geometry has great importance for the theoretical prediction of the Judd-Ofelt intensity parameters, because such parameters depend on the chemical environment around the lanthanide ion (first coordination sphere). To do this, LUMPAC contains a module designed to create a graphics interface with the well-known semiempirical package, MOPAC [1] (Figure 1). This module aims to further facilitate the application of the Sparkle models [2-6], which were developed by our group, and are implemented in the MOPAC program.
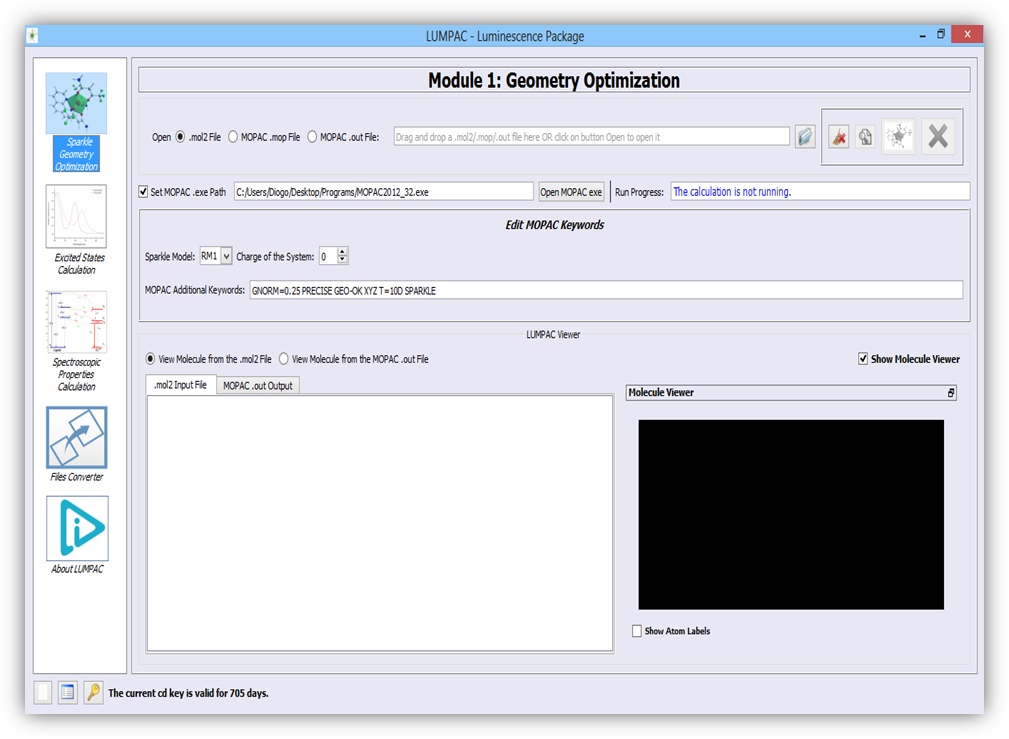
Figure 1. Module designed for the process of geometry optimization using the Sparkle models implemented in the MOPAC computational chemistry package
All features implemented in the LUMPAC program will be demonstrated by using the Eu(DSACAC)3(phen) system (Figure 2) as a case study; where DSACAC stands for 1,5-styrylacetylacetone, and phen stands for phenanthroline [7].
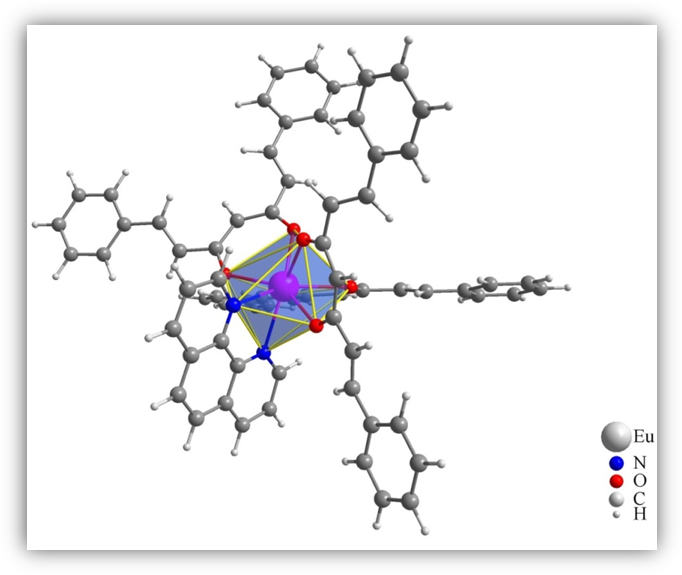
Figure 2. Representation of the molecular structure of the Eu(DSACAC)3(phen) system. The europium coordination polyhedron is highlighted.
A .mol2 file of the desired system is mandatory in order to be able to carry out the geometry optimization using LUMPAC. The .mol2 file can be created by using graphics programs, such as HyperChem [8], Gabedit [9], Avogadro [10], among others. These programs enable the drawing of 3D chemical structures in an interactive manner. A detailed tutorial showing how to build 3D chemical structures using either Gabedit can be accessed at the following link: http://www.sparkle.pro.br/tutorial/drawing-complexes. Moreover, this same link also presents a tutorial on how to draw 3D chemical structures using HyperChem.
Procedure for Geometry Optimization using LUMPAC
- Check if the option "Open .mol2 File" is selected. After that, click on button
 (Figure 3) to open the .mol2 file;
(Figure 3) to open the .mol2 file;
Figure 3 shows the features of each graphic element of the LUMPAC interface in detail. Emphasis is given on the types of files that can be used as input files.
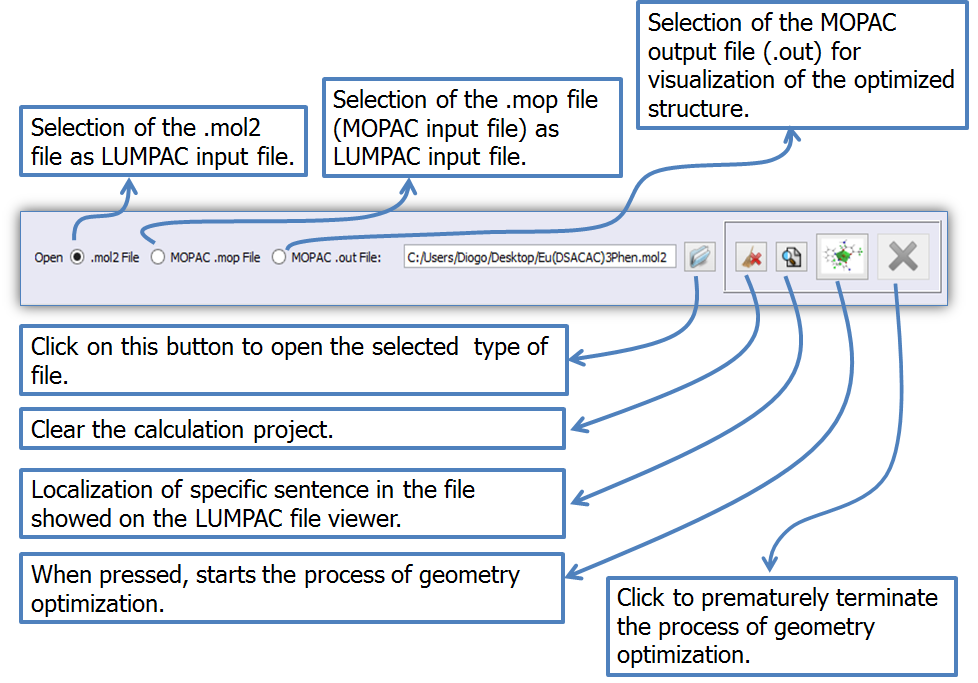
Figure 3. LUMPAC interface showing the types of files that can be used as input files for geometry optimization.
The .mol2 file ("Tripos Mol2 file") is a file format in computational chemistry that contains atom positions in Cartesian coordinates and information on the bonds connecting the atoms. Figure 4 presents the .mol2 file of the Eu(DSACAC)3(phen) system. The coordinates showed in Figure 4 were created by using the Gabedit program, and were previously optimized by using molecular mechanics (AMBER model).

Figure 4. .mol2 file of the Eu(DSACAC)3(phen) system created by Gabedit program.
Attention: It is very important to ensure that, in the drawn structure, all coordination bonds involving the lanthanide ion appear explicitly as shown in Figure 5.
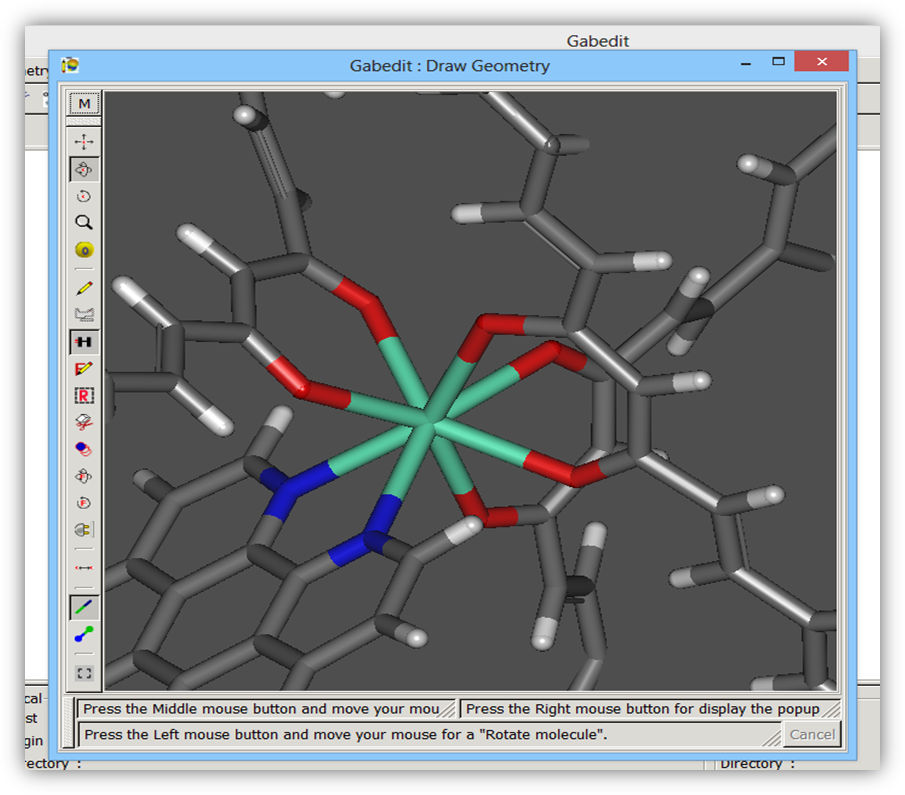
Figure 5. Structure of the Eu(DSACAC)3(phen) complex drawn with Gabedit program showing explicitly all coordinating bonds of the lanthanide ion.
- Click on button
 (Figure 6) to define the external MOPAC program path;
(Figure 6) to define the external MOPAC program path;
LUMPAC program will be distributed with the public domain version of the MOPAC program (version 7.1). Tests showed that this version is quite consistent in the treatment of system containing up to 150 atoms. This way, the public domain version of MOPAC is adequate for studying systems of moderately small size. Others tests carried out showed that the public domain version has some convergence problem in modeling systems with more than 300 atoms. Furthermore, only Sparkle/AM1 [2, 11-15], and Sparkle/PM3 [3, 16-20] are implemented in this version.

Figure 6. LUMPAC interface showing how to integrate an external MOPAC executable into LUMPAC.
MOPAC2012 is freely available at http://openmopac.net. The license for using MOPAC can be requested through of the link http://openmopac.net/download-c.html by filling out a simple form or directly to prof. Stewart, creator and developer of MOPAC, via email (MrMOPAC@OpenMOPAC.net). MOPAC2012 contains all Sparkle models developed so far (Sparkle/AM1 [2, 11-15], Sparkle/PM3 [3, 16-20], Sparkle/PM6 [4], Sparkle/PM7 [5], and Sparkle/RM1 [6]. Due to the stability in geometry optimizations of larger systems, as well as the greater number of available methods, it is strongly recommended the usage of MOPAC2012.
The integration of MOPAC to LUMPAC is quite simple. It is only necessary to provide the path where MOPAC is located (Figure 6).
- The keywords (Figure 7) must be adequately specified before performing the geometry optimization As soon as the .mol2 file is loaded, the button
 (Figure 3) will be activated;
(Figure 3) will be activated;
The keywords are edited by using the LUMPAC interface (Figure 7). Important parameters are: the type of method to be used, and the total charge of the system. The line edit automatically completes the keywords as they are typed. This way, it ensures the user enters the words with the correct syntax.

Figure 7. Editor of MOPAC keywords in geometry optimization module.
- Click on button to run the geometry optimization calculation using the MOPAC.
Attention: the output file generated by MOPAC will have the file extension .out and the same file name of the input file. This output file will be saved in the same directory of the input file. As the MOPAC output file undergoes changes during the program run, it will be updated in the LUMPAC file viewer, providing information about the process of geometry optimization.
- When the calculation is finished, the user can choose to visualize either the starting structure, or the optimized structure (Figure 8).
Figure 8 shows the file and molecule viewers of LUMPAC in the geometry optimization module in detail. It is possible to highlight the molecule viewer if the user wants to visualize the molecular structure in detail (Figure 9). To return the molecule viewer to its initial position, double click it.
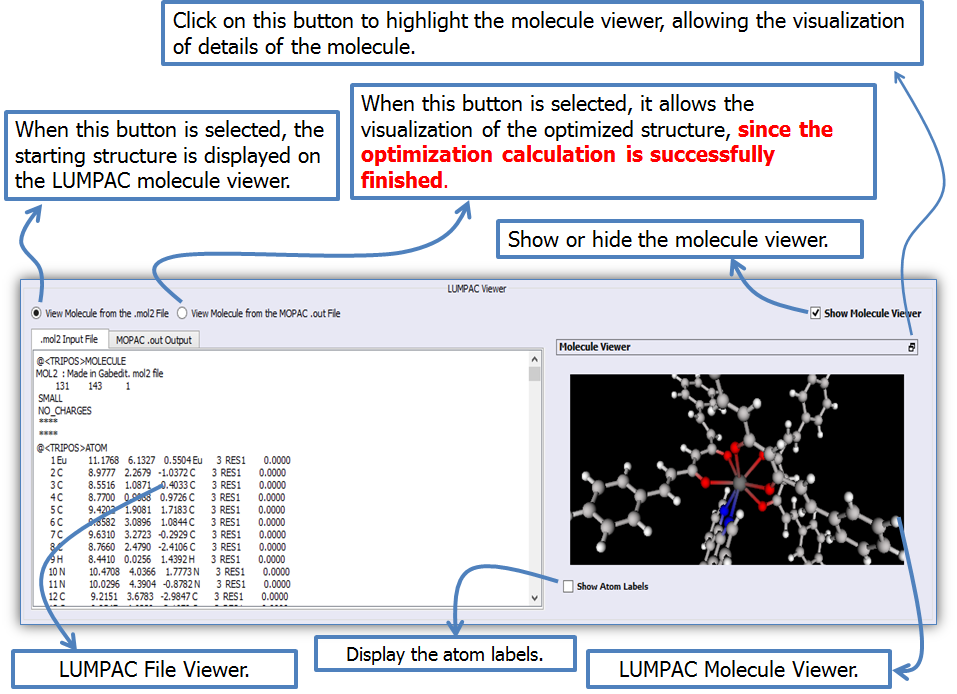
Figure 8. File and molecule viewers of LUMPAC in the geometry optimization module
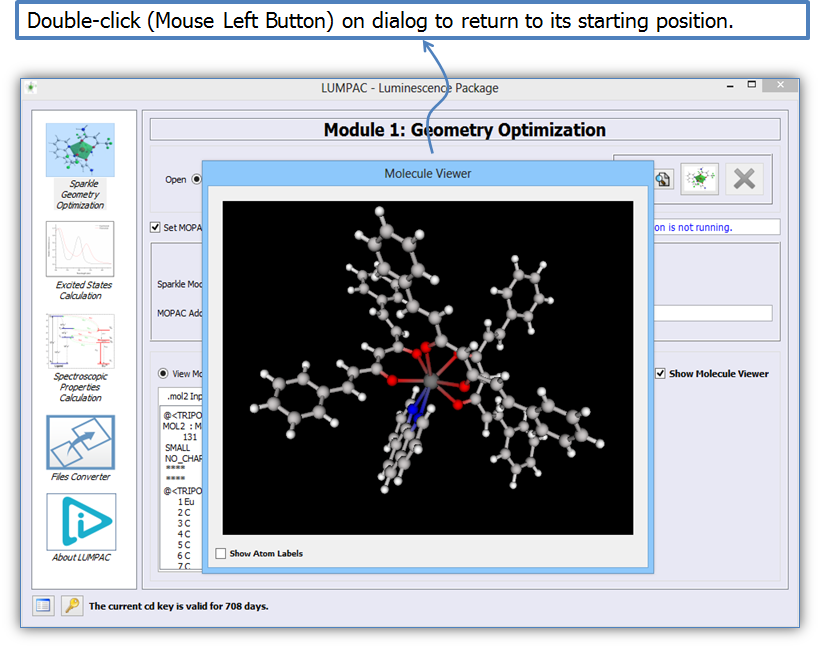
Figure 9. Visualization of the Eu(DSACAC)3(phen) system optimized with the Sparkle/RM1 model.
The visualization configuration can be changed by using the mouse. Table 1 shows the commands related to the translation, rotation, and zoom functions.
Table 1. Commands related to the translation, rotation, and zoom functions of the visualized structure (molecule viewer).
| Commands | Function |
|---|---|
| Left button pressed and moving the mouse. | The visualization is freely moved. |
| SHIFT + Left button pressed and moving the mouse. | A translation is executed. |
| Right button pressed and moving the mouse. | By moving the mouse forward, the zooming is increased; it is reduced by moving it backwards. |
| (i) Mouse scroll wheel, or (ii) by pressing the right mouse button and moving the mouse either forwards (to increase the zooming) or backwards (to decrease the zooming). | The zooming is changed. |
| CTRL + Left button pressed and moving the mouse. | The visualization is rotated. |
LUMPAC can run the geometry optimization calculation using a .mop (MOPAC input file) file as a LUMPAC input file. When this option is selected, it is not allowed to edit the MOPAC keywords using the LUMPAC interface. If the user wants to change the keywords of a .mop file, it will be necessary to open it by using a text editor for editing it.
Bibliographic References
[1] Stewart, J.J.P., MOPAC2012, 2012, Stewart Computational Chemistry, CO, Colorado Springs: USA.
[2] Freire, R.O., G.B. Rocha, and A.M. Simas, Sparkle model for the calculation of lanthanide complexes: AM1 parameters for Eu(III), Gd(III), and Tb(III). Inorganic Chemistry, 2005. 44(9): p. 3299-3310.
[3] Freire, R.O., G.B. Rocha, and A.M. Simas, Sparkle/PM3 for the Modeling of Europium(III), Gadolinium(III), and Terbium(III) Complexes. Journal of the Brazilian Chemical Society, 2009. 20(9): p. 1638-1645.
[4] Freire, R.O. and A.M. Simas, Sparkle/PM6 Parameters for all Lanthanide Trications from La(III) to Lu(III). Journal of Chemical Theory and Computation, 2010. 6(7): p. 2019-2023.
[5] Dutra, J.D.L., et al., Sparkle/PM7 Lanthanide Parameters for the Modeling of Complexes and Materials. Journal of Chemical Theory and Computation, 2013. 9(8): p. 3333-3341.
[6] Filho, M.A.M., et al., Sparkle/RM1 parameters for the semiempirical quantum chemical calculation of lanthanide complexes. RSC Advances, 2013. 3(37): p. 16747-16755.
[7] Liang, H. and F. Xie, Photoluminescence study of a europium (III) complex containing 1,5-styrylacetylacetone ligands. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 2010. 77(2): p. 348-350.
[8] Hypercube, I., HyperChemTM Professional 8: Gainesville, FL.
[9] Allouche, A.-R., Gabedit—A graphical user interface for computational chemistry softwares. Journal of Computational Chemistry, 2011. 32(1): p. 174-182.
[10] Hanwell, M.D., et al., Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. Journal of Cheminformatics, 2012. 4(1): p. 1-17.
[11] da Costa, N.B., et al., Sparkle/AM1 modeling of holmium (III) complexes. Polyhedron, 2005. 24(18): p. 3046-3051.
[12] Freire, R.O., G.B. Rocha, and A.M. Simas, Modeling lanthanide complexes: Sparkle/AM1 parameters for ytterbium (III). Journal of Computational Chemistry, 2005. 26(14): p. 1524-1528.
[13] Freire, R.O., et al., Modeling lanthanide coordination compounds: Sparkle/AM1 parameters for praseodymium (III). Journal of Organometallic Chemistry, 2005. 690(18): p. 4099-4102.
[14] da Costa, N.B., et al., Sparkle model for the AM1 calculation of dysprosium (III) complexes. Inorganic Chemistry Communications, 2005. 8(9): p. 831-835.
[15] Freire, R.O., G.B. Rocha, and A.M. Simas, Modeling rare earth complexes: Sparkle/AM1 parameters for thulium (III). Chemical Physics Letters, 2005. 411(1-3): p. 61-65.
[16] Freire, R.O., G.B. Rocha, and A.M. Simas, Modeling rare earth complexes: Sparkle/PM3 parameters for thulium(III). Chemical Physics Letters, 2006. 425(1-3): p. 138-141.
[17] Freire, R.O., et al., Sparkle/PM3 parameters for the modeling of neodymium(III), promethium(III), and samarium(III) complexes. Journal of Chemical Theory and Computation, 2007. 3(4): p. 1588-1596.
[18] Freire, R.O., G.B. Rocha, and A.M. Simas, Sparkle/PM3 parameters for praseodymium(III) and ytterbium(III). Chemical Physics Letters, 2007. 441(4-6): p. 354-357.
[19] Simas, A.M., R.O. Freire, and G.B. Rocha, Cerium (III) complexes modeling with Sparkle/PM3. Computational Science - Iccs 2007, Pt 2, Proceedings, 2007. 4488: p. 312-318.
[20] Simas, A.M., R.O. Freire, and G.B. Rocha, Lanthanide coordination compounds modeling: Sparkle/PM3 parameters for dysprosium (III), holmium (III) and erbium (III). Journal of Organometallic Chemistry, 2008. 693(10): p. 1952-1956.