LUMPAC
Lanthanide Luminescence
Software Package
Quick Start Guide
Version 1.4.1
By
Pople Computational Chemistry Laboratory
Departamento de Química
Universidade Federal de Sergipe
São Cristóvão, SE, Brazil
LUMPAC
Powerful, efficient and user friendly
LUMPAC carries out a complete theoretical study of the luminescence of lanthanide containing systems, such as lanthanide-based luminescent hybrid materials, and can also be applied to functional materials and to bio-sciences. LUMPAC consists of four modules:
Module #1:
Optimizes the geometries of lanthanide containing systems such as complexes, solids, MOFs, for any of the fifteen lanthanide trications, from La(III) to Lu(III), with either RM1 or the Sparkle Models, within MOPAC.
Module #2:
Obtain singlet and triplet excited state energies for the lanthanide containing systems, from INDO/S-CIS ORCA calculations.
Module #3:
From the experimental emission spectrum, calculate:
- Chromaticity diagram (CIE 1931 diagram);
- Judd-Ofelt intensity parameters;
- Radiative and nonradiative emission rates;
- Quantum efficiency.
From theoretical photophysics calculations, obtain:
- Judd-Ofelt intensity parameters;
- Energy transfer and back transfer rates;
- Distance from the donor to the acceptor centers of the energy transfer process;
- Radiative and nonradiative emission rates;
- Quantum efficiency;
- Overall quantum yield;
- Chemical partition of the radiative emission rate.
Module #4:
File conversion module – converts files of various formats to one another. Example: from a MOPAC output file, it generates an ORCA input file for INDO/S-CIS calculations, etc.
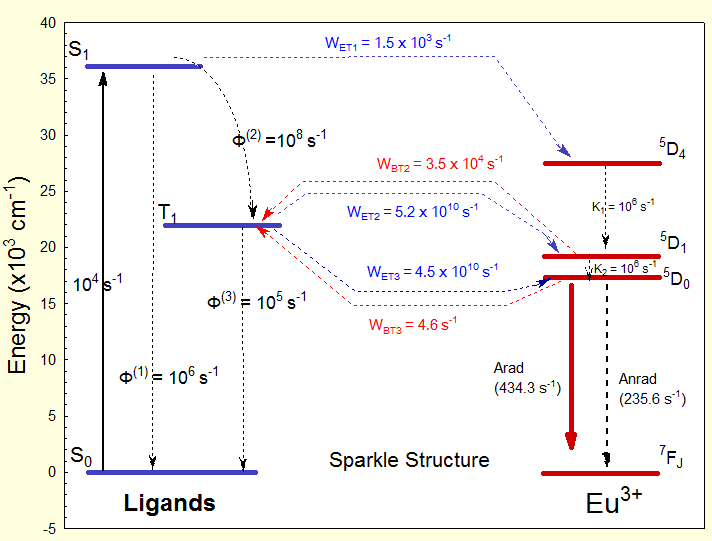
Jablonksi Diagrams
From LUMPAC outputs, and your favorite drawing software, you will be able to create Jablonski diagrams such as the one below:
A Typical Problem
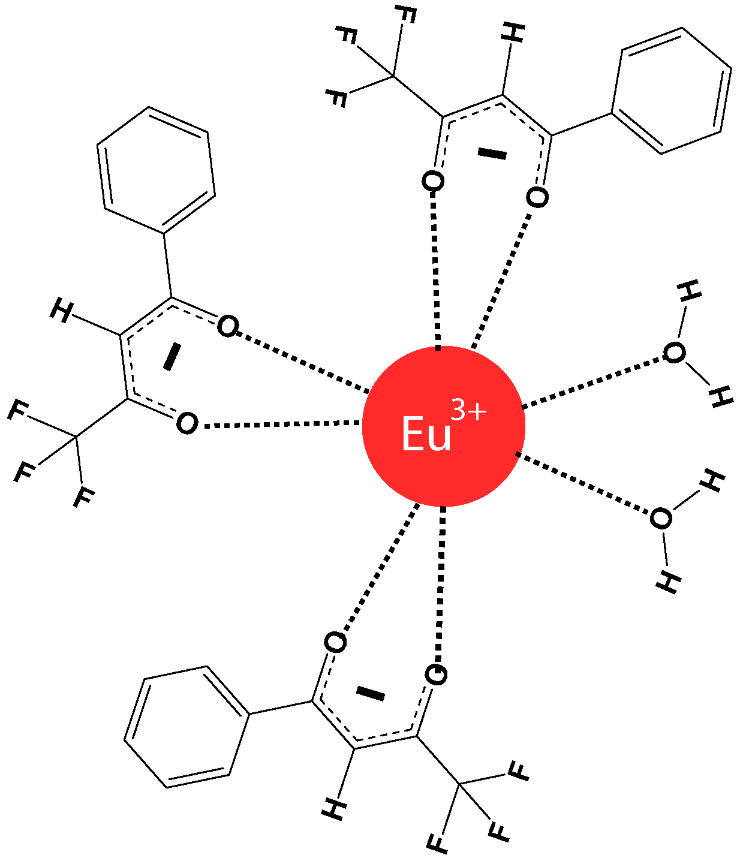
John is a graduate student who tried to reproduce the work of Nathalia B. D. Lima, Simone M. C. Gonçalves, Severino A. Júnior, and Alfredo M. Simas, published in Scientific Reports, 2013, 3, p. 2395 http://www.nature.com/articles/srep02395.
He synthesized the europium complex Eu(BTFA)3(H2O)2, which has the following chemical structure:
Subsequently, he measured the emission spectrum of this complex, as below:
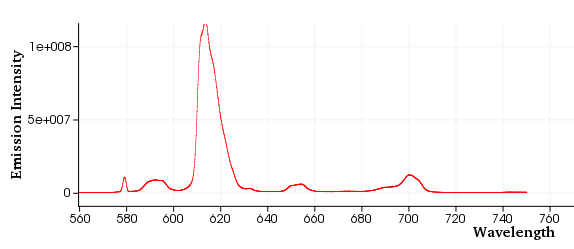
After measurement, the spectrum is usually provided by the equipment as a file with extension .OPJ, such as emission.OPJ, which can be opened with the graphical software Origin® 2016.
Once John opened the .OPJ file in Origin® 2016, two columns appeared: the first with the wavelengths, and, the second, with the emission intensities, as shown below:
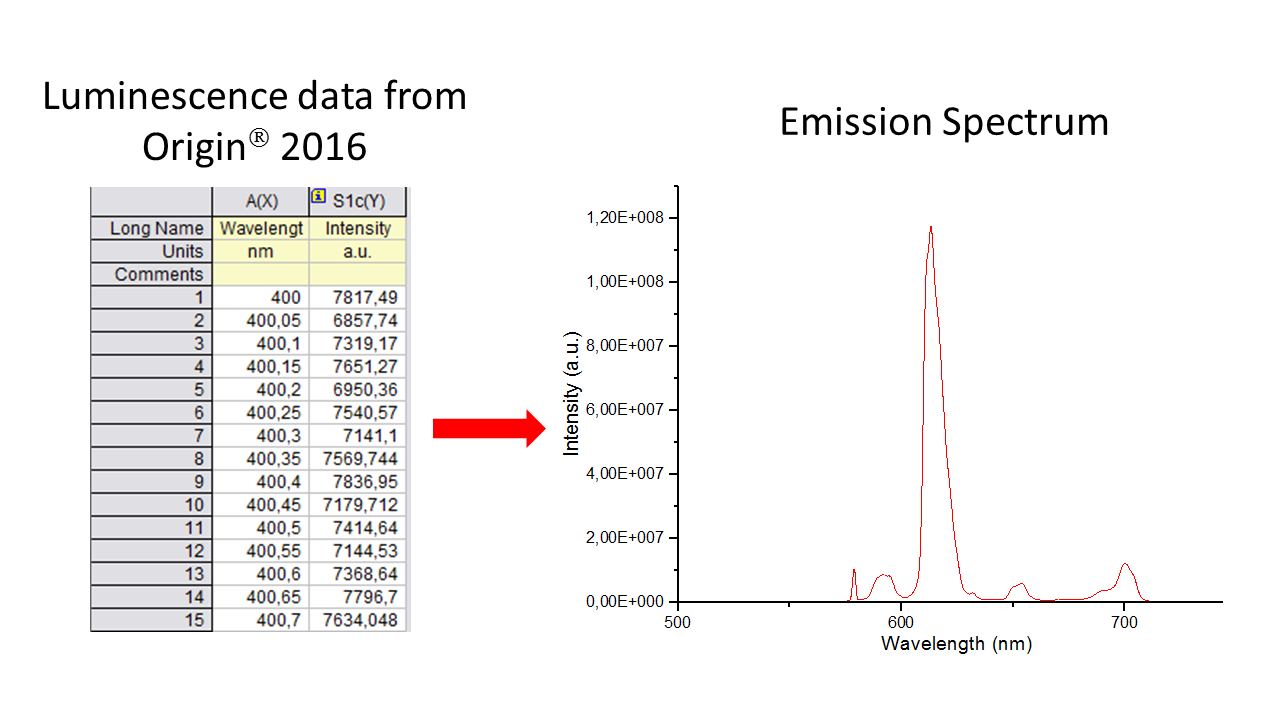
In order to process the data quickly and easily, John used Textpad, which as a very easy and convenient text editor . So, John highlighted the two columns in Origin® 2016, pressed the right button of his mouse and copied the content of both columns. Then he opened Textpad and pasted the two columns. If the decimal mark appears as a comma, it must be replaced by a dot, because LUMPAC uses dots, in which case the F8 key can be pressed to do the job.
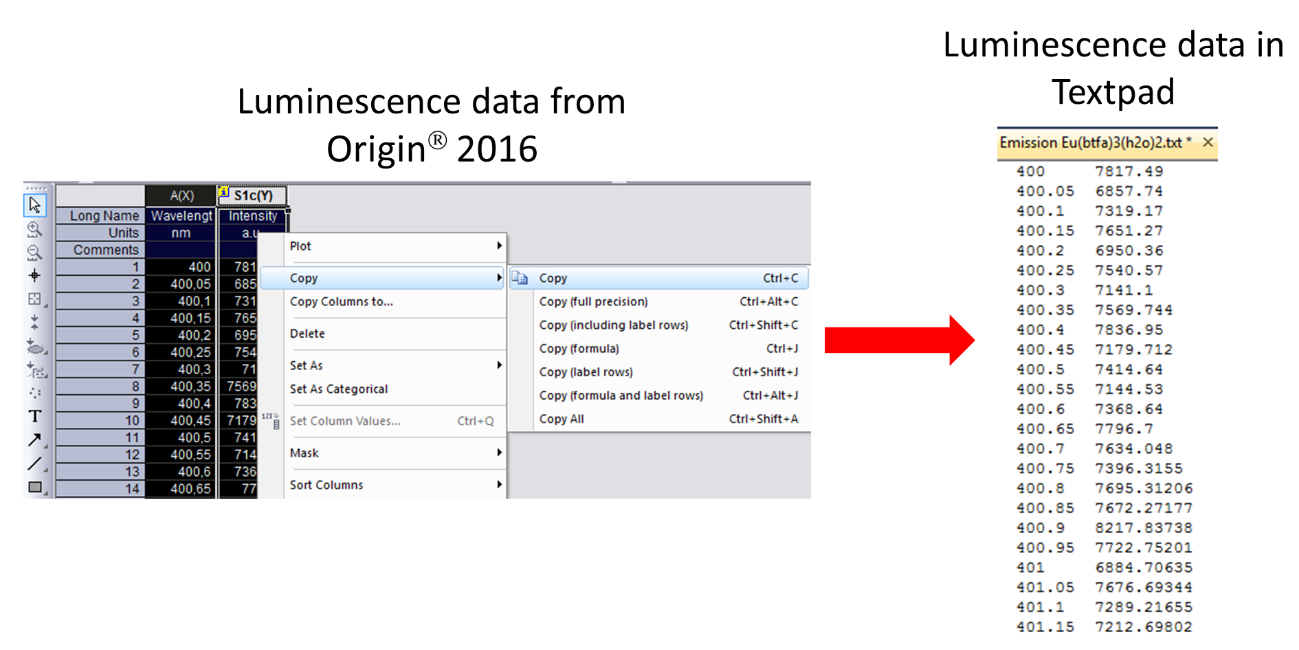
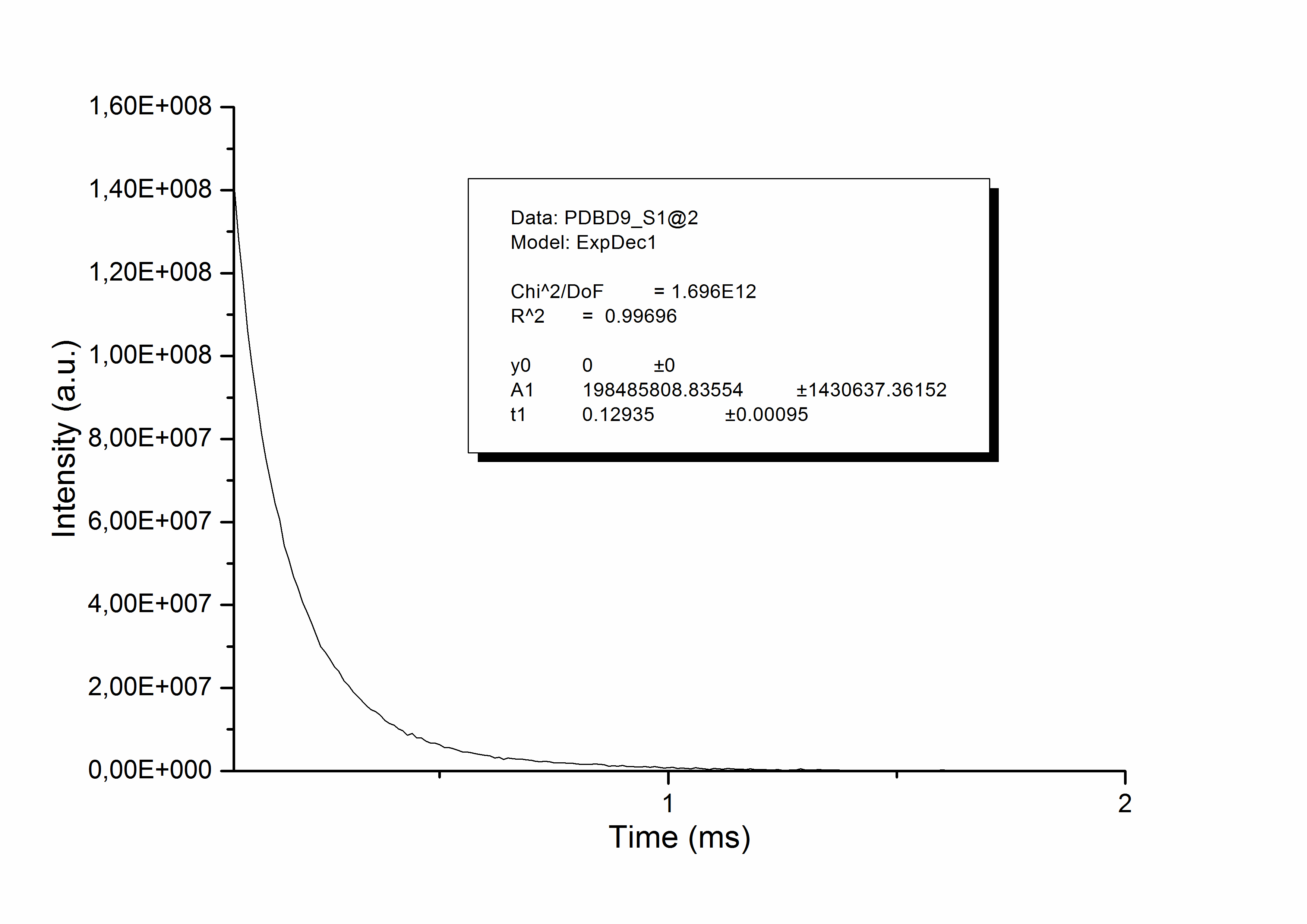
John, then did not need to do anything further, and simply saved the file as a .txt file: emission.txt. This is the file that will be used in LUMPAC. He also measured the experimental lifetime of the emission 5D0 7F2 for this complex as being 0.129ms.
Now, John is in possession of all experimental data needed to start using LUMPAC: file emission.txt, and the measured lifetime.
John now wants to know what can LUMPAC do for him! He visited the LUMPAC site, downloaded LUMPAC and this Quick Start Guide, and decided to give LUMPAC a try!
When John clicked on the LUMPAC executable, a small window opened requesting a key:
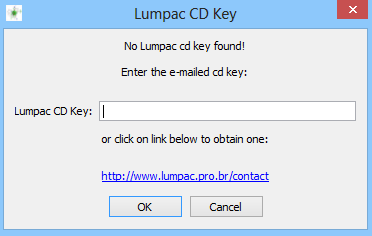
He then clicked on the link, and was sent to the license request page, which he filled promptly:
He clicked on the link, and the e-mail request was sent. He understood that the license would take a while to be processed.
He then proceeded with downloading the other necessary softwares that are required to run LUMPAC, that are MOPAC and ORCA.
MOPAC
MOPAC2016 is freely available for academics at http://openmopac.net. The license for using MOPAC can be requested through the link http://openmopac.net/download-c.html by filling out a simple form or directly to prof. Stewart, creator and developer of MOPAC, via email (MrMOPAC@OpenMOPAC.net). The license usually arrives within one or two days.
John found the instructions on how to install MOPAC in a .txt file that comes bundled with the downloaded compressed file.
Observation: The Installation of MOPAC sometimes can be a little tricky, because it must be performed with administrator privileges - please, pay extra attention to item 2.3 of the installation file named “installation instructions.txt”.
ORCA
The ORCA binary files are freely available for academics. To download them, it is only necessary to fill a form at the following site: http://orcaforum.kofo.mpg.de; no license is needed. Please, be aware that the ORCA installation does not create a shortcut file. When the download ends, you will have in your computer the compacted file containing around 1.5 gigabytes and more than 30 binary .exe files, which must have been extracted to the same directory.
ATTENTION: LUMPAC runs ORCA by command line, and ORCA does not work correctly when its directory name contains any blank character. Thereby, to avoid any problem, it is very important to place the ORCA program in a directory without blank characters in its directory name. We strongly recommend that you use C:\orca.
John, then proceeded with the installations of ORCA and MOPAC according to the instructions on the respective sites and on the e-mail from MrMOPAC.
LUMPAC
To his surprise, on the next day, the e-mail with the LUMPAC license arrived. It was a 29 digit alphanumeric code such as this one: 000000290801000102234257EX354 He clicked on the LUMPAC executable and, this time, he entered the license key!
Module 1: Geometry Optimization
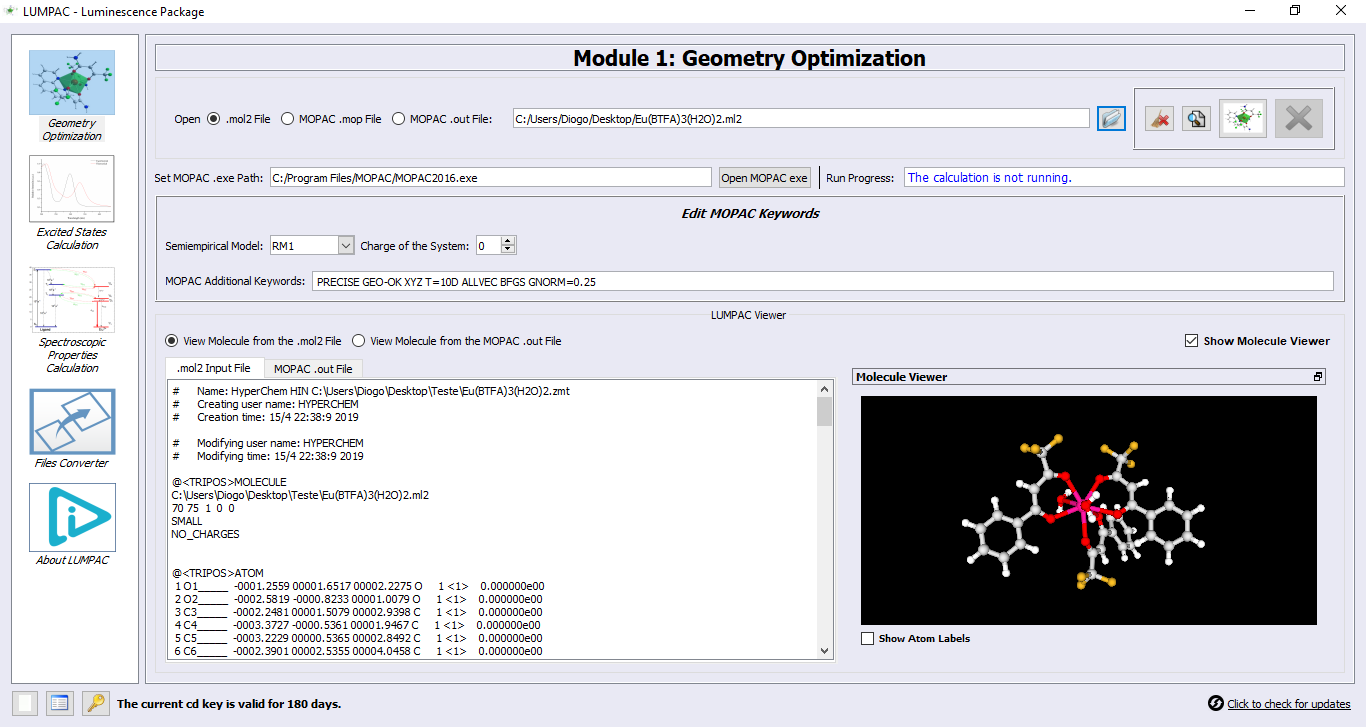
1. John then prepared the starting geometry of the complex using standard molecular modeling softwares such as Hyperchem (recommended and available from http://www.hyper.com/) or the free software GABEDIT (a bit more difficult to use). Tutorials are available from:
http://www.sparkle.pro.br/tutorial/drawing-complexes.
If John has the .cif file corresponding to his compound, by using LUMPAC, he will be able to calculate all spectroscopic properties from the experimental crystallographic geometry. But in order to do this, he will need to first convert the .cif to a .mol2 file by using, for example, the MERCURY program. Moreover, John must not forget to add the keyword 1SCF in the “MOPAC Additional Keywords” field. By using the keyword 1SCF, MOPAC will not change or attempt to optimize the starting crystallography geometry. From the input .mol2 file, a MOPAC output file will be generated containing the Cartesian coordinates, reordered in a convenient manner.
2. Click on button  and define the external MOPAC program path;
and define the external MOPAC program path;
3. Make sure that the correct structure file option is selected. Then, click on button  to open, for example, the .mol2 file with the structure of the complex that John had already prepared:
to open, for example, the .mol2 file with the structure of the complex that John had already prepared:
4. As soon as the MOL2 file is loaded, the button  will be enabled. Pressing it, executes the MOPAC geometry optimization;
will be enabled. Pressing it, executes the MOPAC geometry optimization;
5. Attention: the output file generated by MOPAC will have the file extension .out and the same file name of the input file. This output file will be saved in the same directory of the input file. As the MOPAC output file undergoes changes during the program run, it will be updated in the LUMPAC file viewer, providing information about the process of geometry optimization;
6. When the calculation is finished, the user can choose to visualize either the starting structure, or the optimized structure:

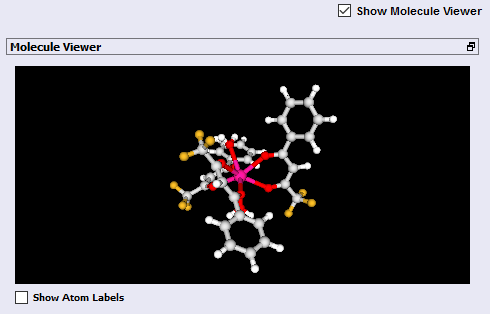
John now has a Sparkle Model optimized geometry, and is ready to go to Module 2!
Module 2: Excited States Calculation
John, pressed the Module 2 icon button, as indicated by the arrow:
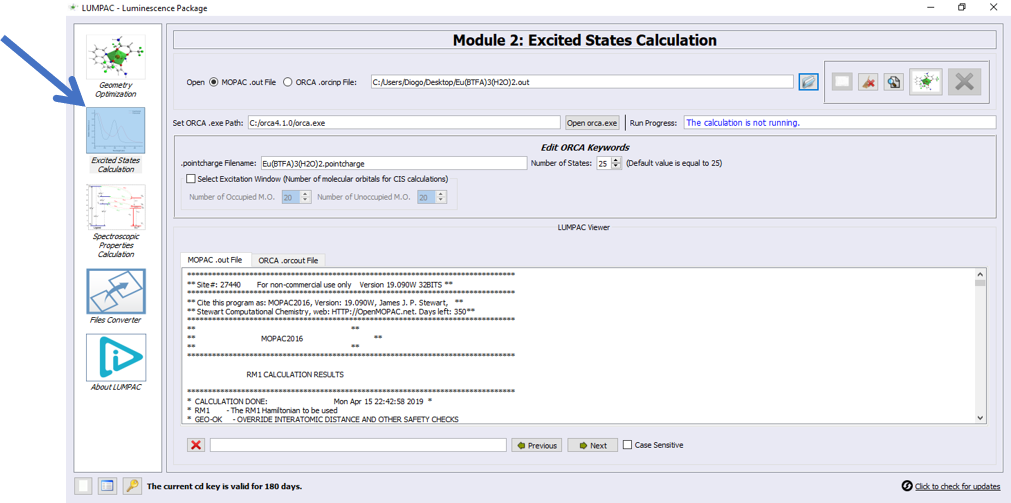
1. Click on button  and define the directory path of the external ORCA program;
and define the directory path of the external ORCA program;
2. Click on button to open the MOPAC output file (.out) created by the process of geometry optimization described in Module 1;
3. Button will be enabled as soon as the MOPAC .out file is loaded. Click on it to run the ORCA calculation;
4. The ORCA output file will have the same name of the .orcinp file, but with a different file extension (.orcout).
John now has the energies of the excited states of his complex, and is now ready to go to Module 3.
Module 3: Spectroscopic Properties Calculation
John, pressed the Module 3 icon button, as indicated by the arrow, to go to Module 3:
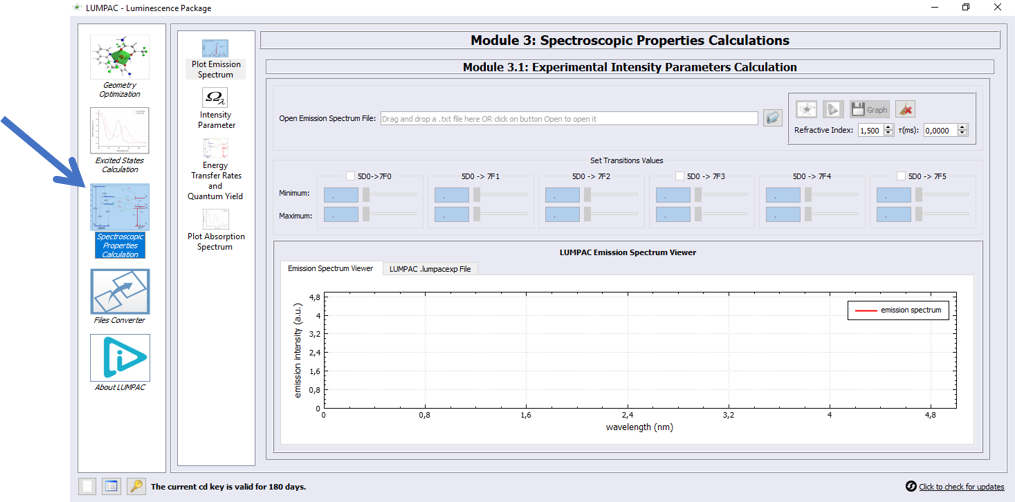
1. Click on button to open the .txt file of the emission spectrum file containing the wavelengths versus emission intensities;
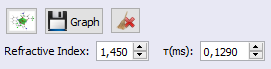
2. Provide the experimental lifetime to calculate the non-radiative emission rate to John's value of 0.1290ms and also the refractive index of the medium, in this case 1.450 since John carried out his experiments in chloroform solution, as shown below:
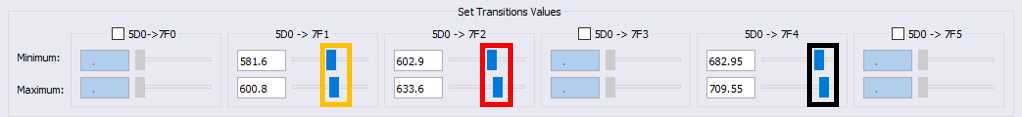
3. The areas related to the 5D0 → 7F1 , 5D0 → 7F2 and 5D0 → 7F4 transitions must be adequately chosen by using the LUMPAC interface.
Using the slider  select the wavelength range under each of the main peaks of the emission spectrum so that LUMPAC can integrate the respective areas.
select the wavelength range under each of the main peaks of the emission spectrum so that LUMPAC can integrate the respective areas.
For example, for the 5D0 → 7F1 transition, use the orange slider (circled in orange in the figure below). Be sure to select all three spectral ranges, as in the figure below.
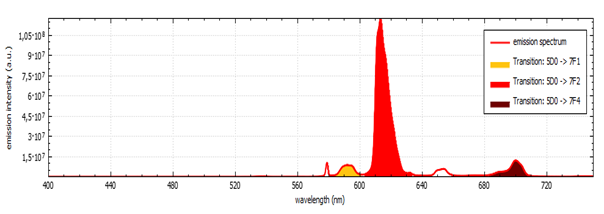
As you choose the wavelength intervals, the areas under the curves will be painted, as shown below. As you choose the wavelength intervals, the areas under the curves will be painted, as shown below.
4. Click on button to start the luminescence properties calculation;
5. Click on button  to obtain the chromaticity diagram corresponding to the provided emission spectrum;
to obtain the chromaticity diagram corresponding to the provided emission spectrum;
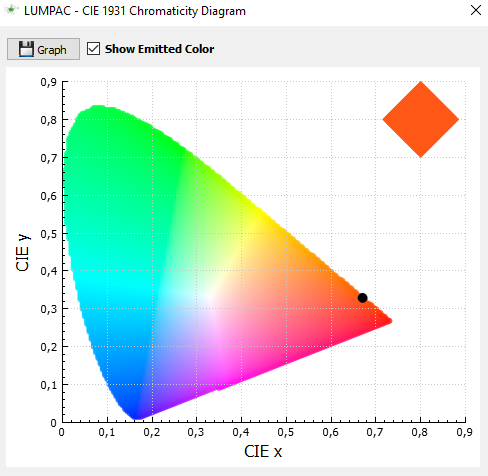
John now has the experimental intensity parameters, and is now ready to go calculate the theoretical luminescence properties.
Theoretical Calculation of the Intensity Parameters
John, pressed the Intensity Parameters icon button, as indicated by the arrow.
1. Click on button to open the MOPAC output file Eu(BTFA)3(H2O)2.out which contains the optimized geometry;
2. Define the same value of refractive index that was used in the last step, as shown in the following image:
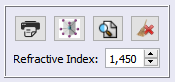
3. Define the coordination number in order to show the ligand atom labels in the molecule viewer. In this case, set the coordination number to 8 using the updown arrows as indicated below.
Please, note that, in LUMPAC, europium must always be the first atom appearing in the structure files (either .mol2, .mop, or .out) followed by all its directly coordinated atoms.
To order the atoms properly, we recommend that, after a MOPAC Sparkle calculation, you select the optimized geometry from the .arc file that was produced (and which always shows the geometry in Cartesian coordinates), and that you reorganize the atoms accordingly, saving the result in a new .mop file, as shown below:
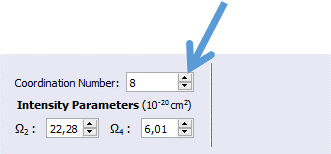
4. In, LUMPAC two different approaches are now available for the adjustment procedure needed for the calculation of the theoretical intensity parameters. The default one has been recently published and is called QDC and provides unique values for charge factors and polarizabilities that, in turn, provide a unique set of intensity parameters for a given geometry. To use this new approach, it is very important that the keyword ALLVEC be specified in the MOPAC geometry optimization calculation . From the Sparkle wavefunction, the electronic densities and Simas Superdelocalizabilities are calculated: both are used in the QDC adjustment. Differently from the previous and now obsolete adjustment scheme, in QDC one does not need to select the atoms that are forced to have the same charge factors and polarizabilities. John decided to choose the QDC adjustment as shown below.
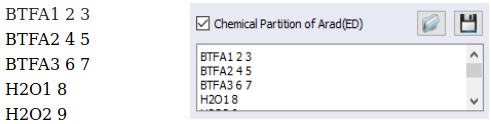
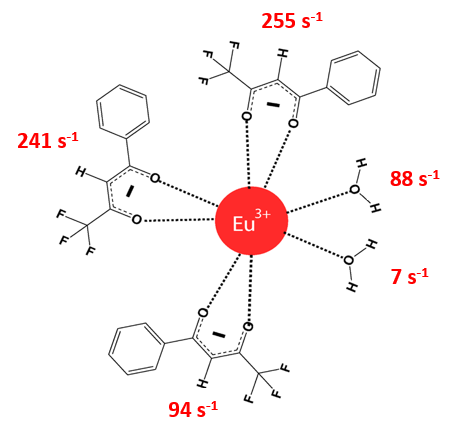
5. John also decided to enable the chemical partition in order to better interpret the effects of the ligands on the electric dipole radiative decay rate Arad(ED), which is defined as a sum of the radiative decay rates of the transitions from 5D0 → 7FJ, with J =2,4,6. The chemical partition of Arad(ED) is calculated for each of the coordinated atoms. At this point, it is very useful to organize the coordinated atoms into groups. Any sorting can be defined: (i) grouping by ligand; (ii) grouping by type of ligands (for example, all coordinated atoms from all three b-diketonates bundled together); (ii) grouping by classes of ligands (either ionic or non-ionic ligands); etc.
John will have to use his creativity to arrive at a grouping that will prove the most useful for his purposes. In this case, John decided to group the atoms by ligands. So, by examining the structure of the complex in the viewing window, and by identifying the coordinated atoms that belonged to the same ligand, he defined the groups as:
Where the first 6 characters name the group (and can be anything), and the next numbers, separated by spaces, denote each coordinated atom. John was very careful to (i) not assign the same atom to two different ligands, and (ii) not leave any coordinated atom out; otherwise the sum of the partitions will not be equal to the total Arad(ED).
Likewise, John could have defined the groupings in terms of types of ligands, as well, by
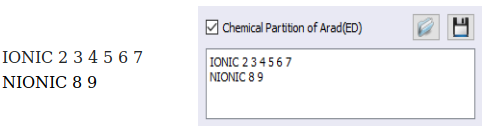
Or, he could also have defined the groupings in terms of classes of ligands as below.
Later, by using a hand calculator or a spreadsheet, John will be able to group the chemical partition by atoms in any way he wishes.
6. Click on button  to perform the calculation of the intensity parameters. At this point, the file with the theoretical intensity parameters will be saved in the same directory of file Eu(BTFA)3(H2O)2.out with the name Eu(BTFA)3(H2O)2.omega.
to perform the calculation of the intensity parameters. At this point, the file with the theoretical intensity parameters will be saved in the same directory of file Eu(BTFA)3(H2O)2.out with the name Eu(BTFA)3(H2O)2.omega.
Energy Transfer Rate and Emission Quantum Yield Calculations
John, pressed the Energy Transfer Rate and Quantum Yield icon button, as indicated by the arrow.
John noted that LUMPAC now calculates the energy transfer rates from the singlet and triplet excited states provided by either the ORCA output file, or by the GAUSSIAN output file (.log file), in which case he had to use the keyword pop=full to print all information on the molecular orbitals that will be needed for the RL calculation. In addiction, LUMPAC has implemented three models to calculate the energy transfer rates, being the model recently published by the group led by Malta the default option.

1. Click on button to open the Eu(BTFA)3(H2O)2.orcout;
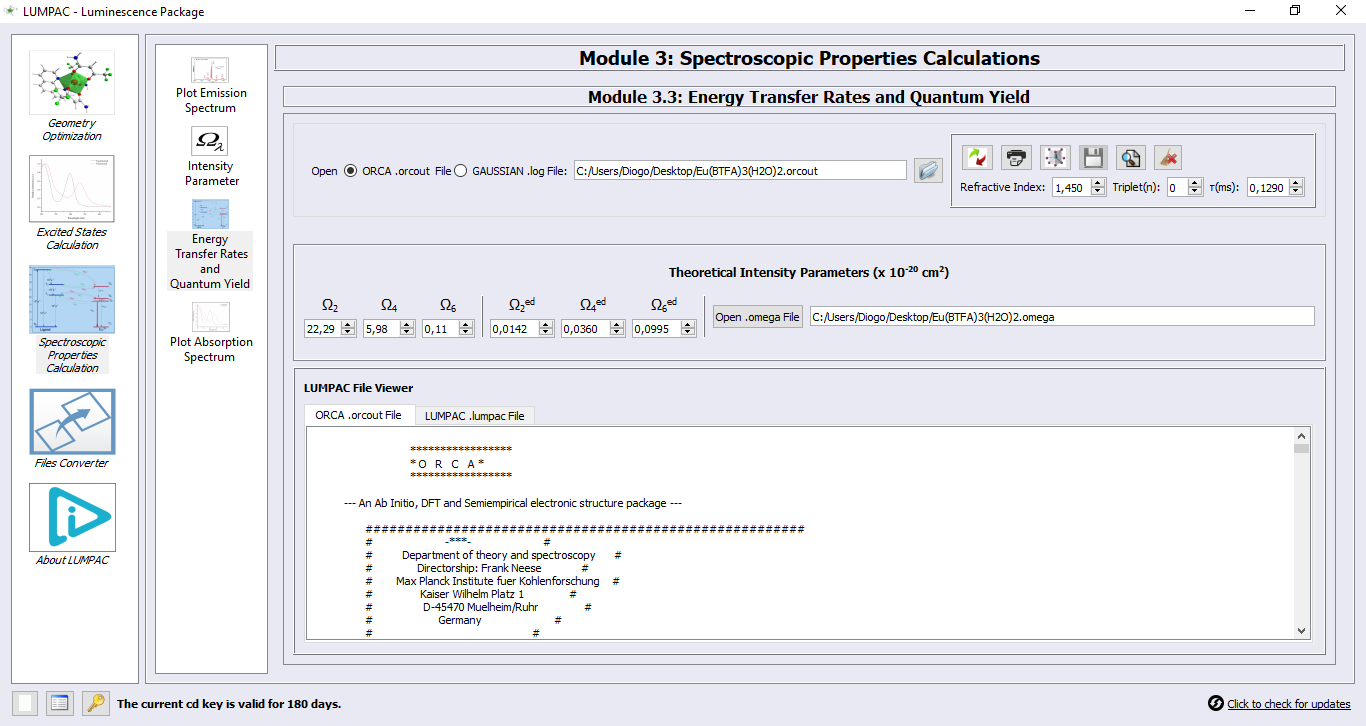
2. Define again the refractive index and the experimental lifetime of the Eu(BTFA)3(H2O)2 complex to 1.450 (chloroform solution) and 0.1290ms, as below;
3. Click on button ![]() to perform the energy transfer and back-transfer rates calculations. Since the experimental lifetime was provided, the overall emission quantum yield will be calculated;
to perform the energy transfer and back-transfer rates calculations. Since the experimental lifetime was provided, the overall emission quantum yield will be calculated;
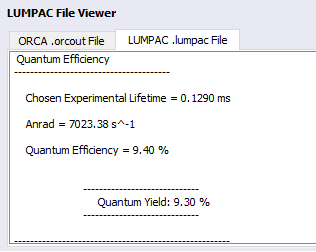
The luminescence properties can all be found, in file Eu(BTFA)3(H2O)2.lumpac. From it, John was able to create the Jablonski diagram! Please, examine this file in detail. An excerpt of this file can be seen below, showing the overall Quantum Yield result:
Theoretical Calculation of the Absorption Spectrum
Finally, John, pressed the Plot Absorption icon button, as indicated by the arrow:
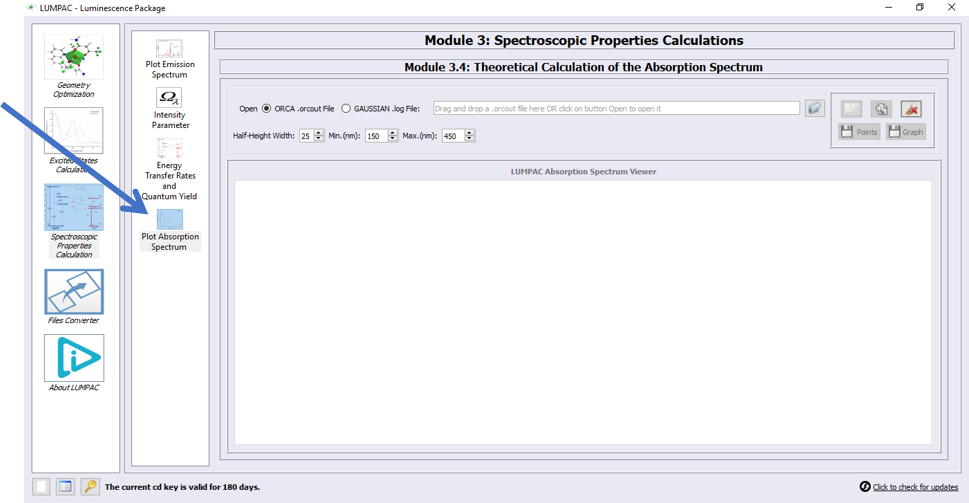
From the GAUSSIAN output file it is also possible to obtain the absorption spectrum.
1. Click on button to open the ORCA Eu(BTFA)3(H2O)2.orcout file;
2. Click on button  to perform the calculation of the absorption spectrum, which will appear as below.
to perform the calculation of the absorption spectrum, which will appear as below.
Wow!! said John.
I will definitely learn all details on how to use LUMPAC; I will read the whole LUMPAC manual; and I will never forget to cite all of the required papers to support the authors!